Introduction
The basic principle of redox flow batteries (RFBs) is to separate two compounds that would otherwise engage in a reaction, so that they remain passive and stable until we allow an electric current to flow. Then the reaction proceeds and electrons are swapped between the compounds, releasing or storing energy according to demand.
Besides the challenges of running a large and complex technical system such as a large-scale RFB (e.g. pumping, heat management, etc.), the choice of the electroactive material is pivotal. Obviously, it should have a high energy and power density. However, to ensure competitive and sustainable energy storage, the material also needs to be widely available, low cost, environmentally safe, and stable over many cycles of charging and discharging.
So far, the materials in use fail us in at least one of these criteria, so the search for new compounds is underway. Organic chemicals represent a huge search space, and given the existence of demonstrators and/or natural redox compounds, we can be confident that plenty of suitable materials exist.
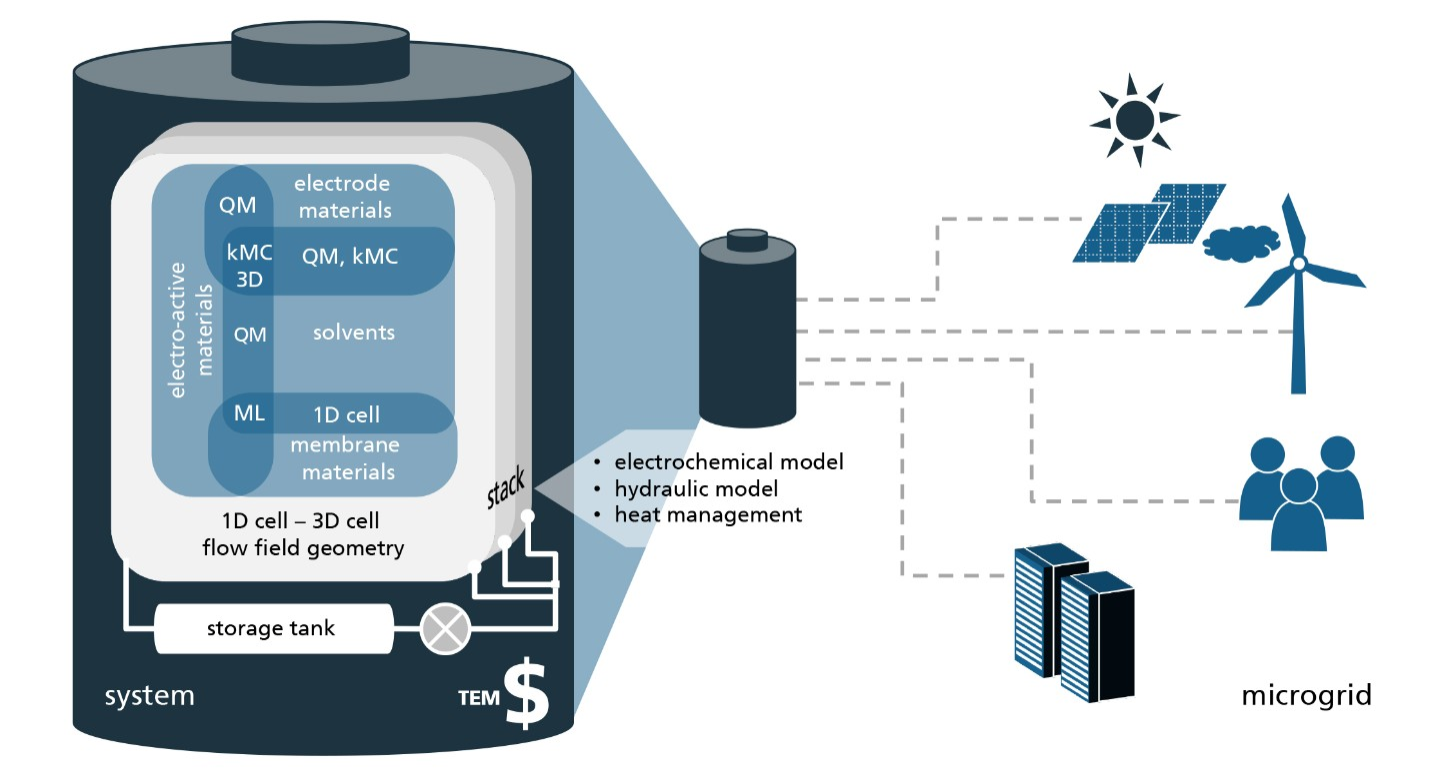
The project SONAR aims to promote the search for new materials by providing a range of computational tools and techniques needed to investigate every aspect of a novel RFB system virtually, i.e. before it enters the stage of costly lab experiments.
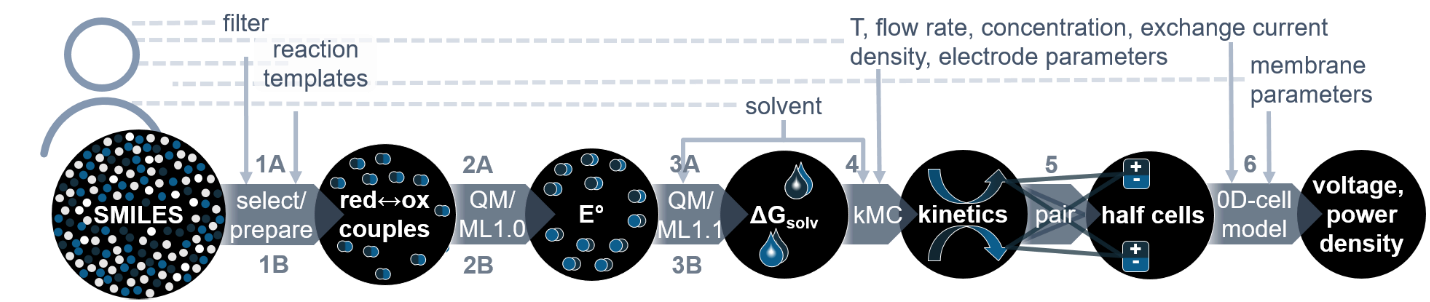
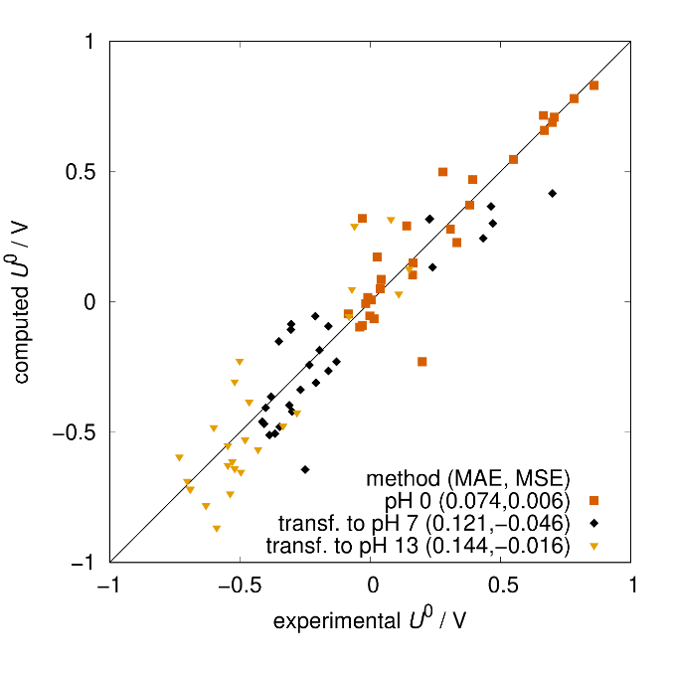
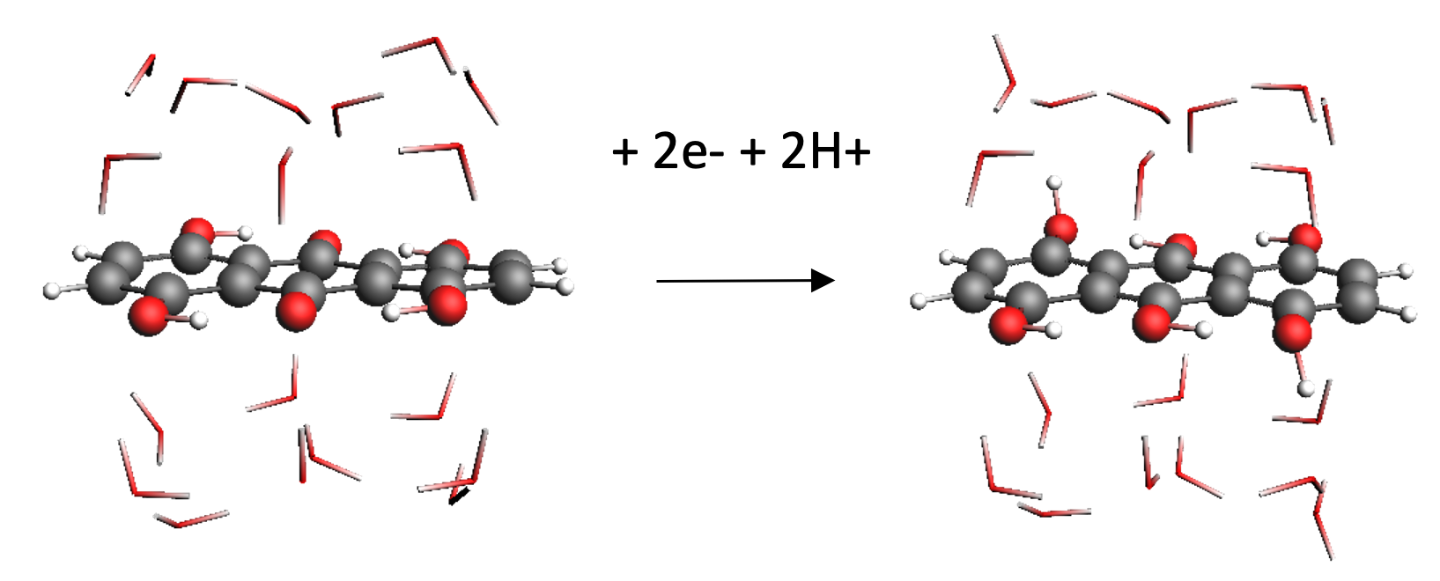
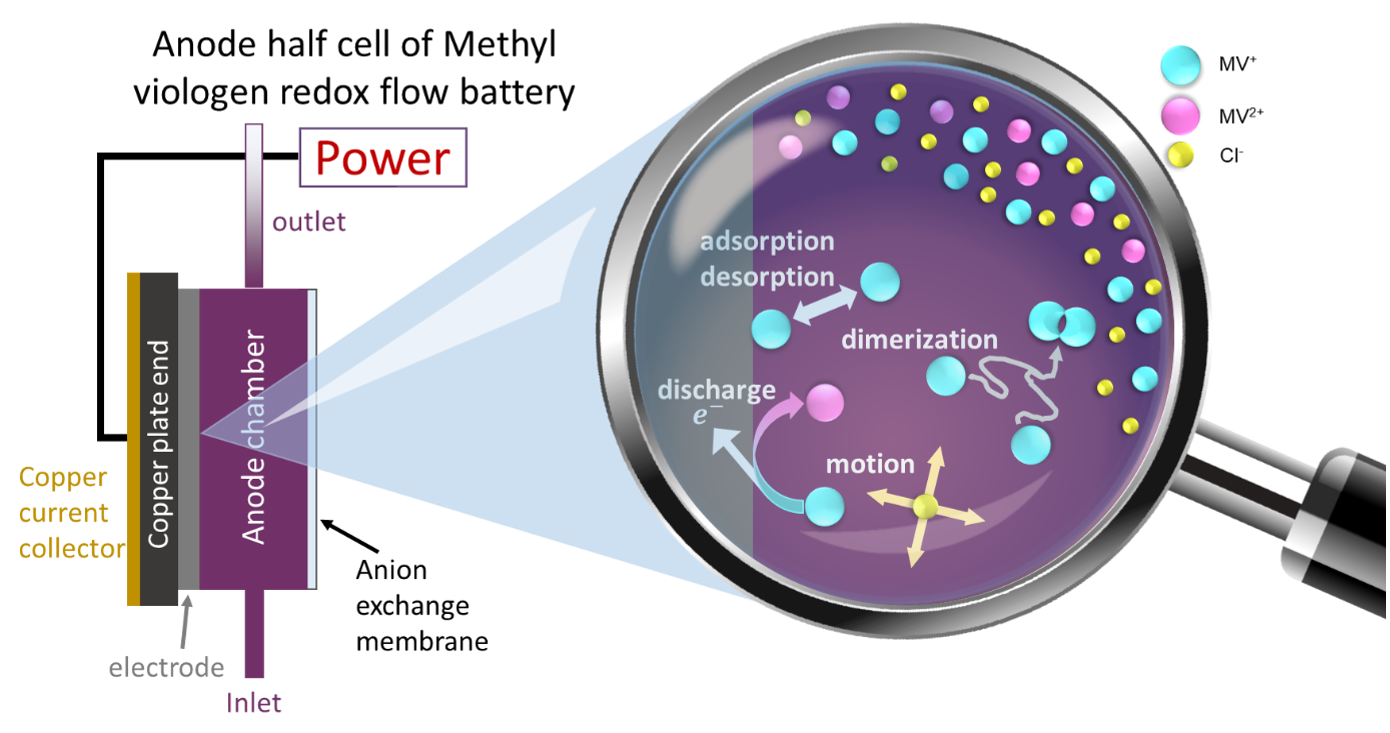
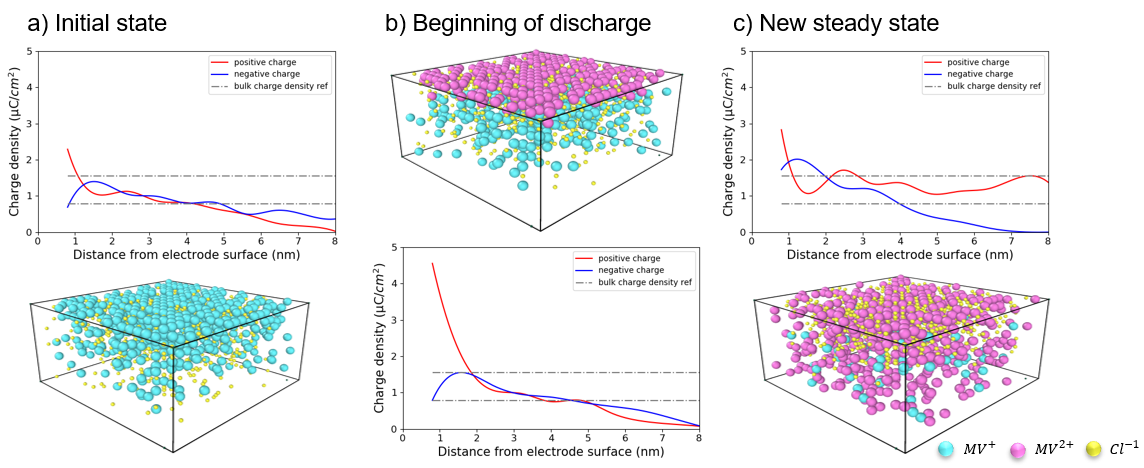
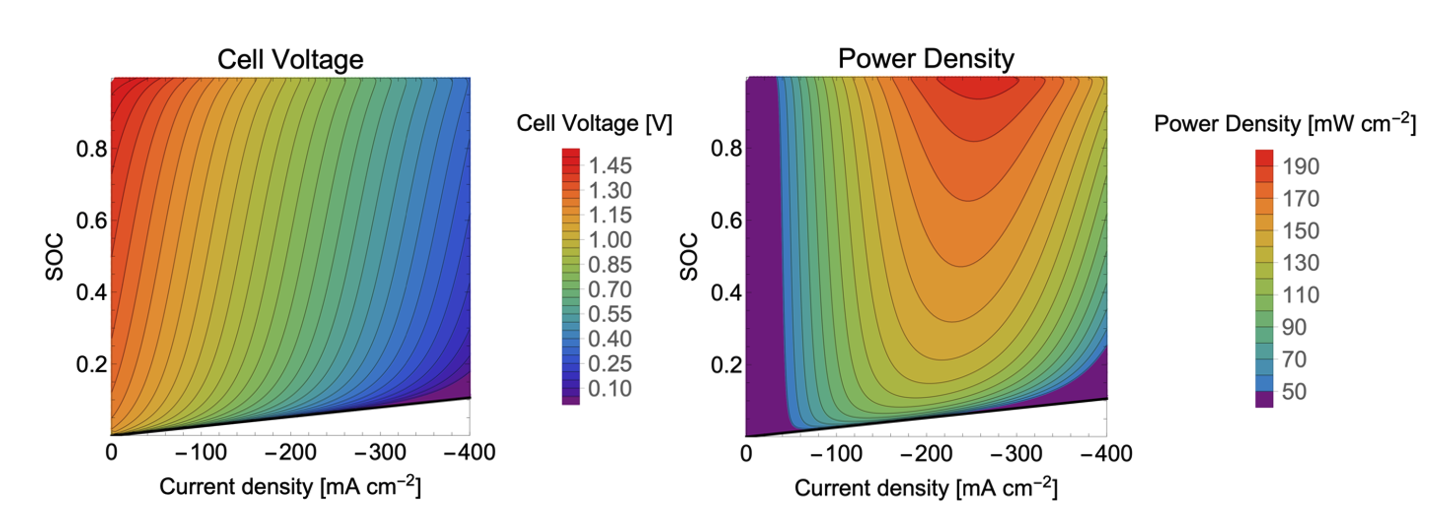
Quantum mechanical calculations give us the theoretical gain in energy for a given redox-reaction, but in practice, we experience losses that reduce the benefit. These are due to additional effects, which we can capture by extending the simulation’s scope step by step, starting from the redox compound and then including the other components of a RFB such as the solvent, processes at the electrode and the membrane. We proceed even further up to the flow field, the organization of the stack and the integration of the RFB within a local micro grid. The final goal is to enable high-volume preselection of potential new organic active materials for redox flow batteries.
With this newsletter we report on the most important results achieved so far in the areas of high-volume preselection, atomistic and meso-scale modelling, and cell-level modelling and simulation.